Remnants of extinct monkeys are hiding inside you, along with those of lizards, jellyfish and other animals. Your DNA is built upon gene fragments from primal ancestors.
Now researchers at the Georgia Institute of Technology have made it more likely that ancestral genes, along with ancestral proteins, can be confidently identified and reconstructed. They have benchmarked a vital tool that would seem nearly impossible to benchmark. The newly won confidence in the tool could also help scientists compute ancient gene sequences and use them to synthesize better proteins to battle diseases.
For some 20 years, scientists have used algorithms to compute their way hundreds of millions of years back into the evolutionary past. Starting with present-day gene sequences, they perform what’s called ancestral sequence reconstruction (ASR) to determine past mutations and figure out the genes’ primal forerunners.
“With the help of ASR, we can now actually build those ancient genes in the laboratory and express their encoded ancient proteins,” said Eric Gaucher, an associate professor at Georgia Tech’s School of Biological Sciences. In a separate project, his lab is computing ancient proteins that were very effective in blood clotting 80 million years ago, in hopes of using them to fight hemophilia today.
That protein comes from a common ancestor humans share with rats.
Time travel substitute
But ASR algorithms have faced logical criticism. Species based on those primal genes are long extinct, and scientists can’t travel back in time to observe mutations that have happened since. So, how can anyone find any physical benchmark to verify and gauge ASR?
A team of researchers led by Gaucher did it by building an evolutionary framework out of myriad mutations. Then they benchmarked ASR algorithms against it – no time machine required. Their results have shored up confidence that the widely used algorithms are working as they should.
“Most of them did a very good job – 98% accurate,” Gaucher said of contemporary algorithms’ ability to compute ancient gene sequences. Their determination of proteins encoded by those sequences was virtually perfect.
Gaucher, research coordinator Ryan Randall and undergraduate student Caelan Radford published their results on Thursday, September 15, 2016, in the journal Nature Communications. Their research has been funded by the NASA Exobiology program, E.I. du Pont de Nemours and Company (DuPont) and the National Science Foundation.
Holographic tree branches
Ancestral sequence reconstruction is like making a family tree for genes.
The many twigs and branches at the treetop would be sequences from species alive today. Shimmying down the tree, called a phylogeny in genetics, you would find their common ancestors, millions of years old, in the lower branches.
There’s a caveat; none of the lower branches exist any longer. They vanished in the extinction of the species bearing those genetic sequences.
ASR computes them back into place using algorithms based on scientific models of evolution. It’s like replacing missing branches with holographic duplicates.
Algorithm horse race
The accuracy of those evolutionary models has been a historic sticking point. And doubts about the algorithms based on them linger in some circles that hold on to an old, tried-and-true algorithm.
So, Gaucher and researcher coordinator Randall pitted the contemporary model-based, or “maximum likelihood,” algorithms in a race against the generic, or “parsimony,” algorithm.
“Parsimony follows the simplest notion of evolution, which is that very little mutation occurs,” Randall said. The models behind contemporary “maximum likelihood” algorithms, by contrast, are laced with filigree, data-packed details.
For the race, Randall made a track of sorts by putting a gene sequence that made a single protein through multiple mutations to construct a real-life phylogeny. She used methods that closely mimicked natural evolution, but that were much, much faster.
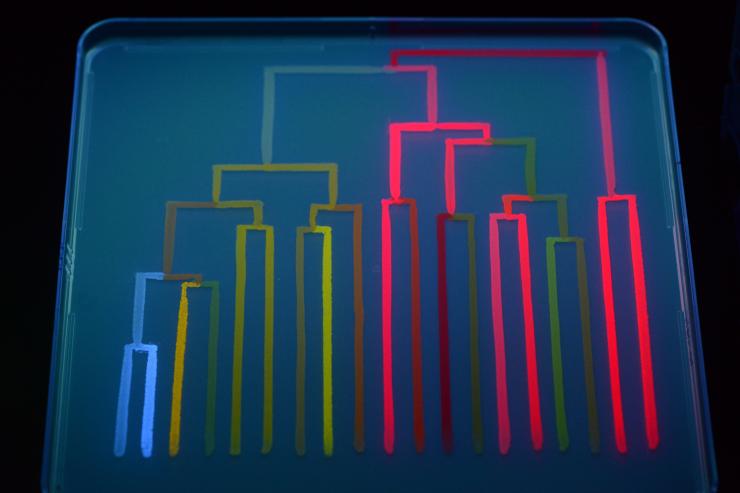
Rainbow phylogeny racetrack
In cells, enzymes called polymerases aid in DNA duplication. They work very efficiently, but their rare mistakes are the most common source of mutations, and Randall took her lead from this.
“We used a polymerase that is error-prone to speed up mutations, and speed up evolution,” she said.
The genes used at the starting point of the lab evolution made a protein that fluoresced red when placed in bacteria. As significant mutations arose, the proteins began changing color. Bacteria containing green fluorescing proteins popped up among the red ones.
Randall divided bacteria with major mutations into new groups, creating branches in the phylogeny, as she went. Many mutations produced new colors – yellow, orange, blue, pink – and Randall ended up with a gene family tree in rainbow colors.
Show me the phenotype
The colors reflected not only new gene sequences but also new phenotypes – the actual proteins they produced, the organism’s working molecules.
“What counts is phenotype,” Gaucher said. “When you analyze DNA strictly by itself, it ignores the context, in which that DNA is connected to phenotype,” he said.
DNA can mutate and still encode the same amino acids, protein’s component parts. Then the mutation has no real effect. But when mutations cause DNA to encode different amino acids, they’re more significant.
A worthy test of ancestral sequence reconstruction algorithms must therefore include phenotype. And Randall took this into account when she selected mutated proteins.
“I selected for variants to purposely make it hard on the algorithms to infer the phenotypes,” she said. The race ensued, and the algorithms got limited information to infer the evolutionary tree’s many dozens of past mutations.
ASR a sure bet
Though the tried-and-true parsimony algorithm performed well, maximum likelihood performed better. “Even though it got the same number of residues (DNA sequences) wrong as parsimony, the incorrectly inferred sequences were still more likely to encode the right phenotypes,” said undergraduate student Caelan Radford, who analyzed the experiment’s statistics.
The margin of error was so tiny that it would not interfere in the determination of past species.
The experiment’s outcome was not too surprising, because prior simulations had predicted it. But the researchers wanted the scientific community to have physical proof that feels trustier than proof from a computer. “It’s a computer algorithm. It will do what you will tell it to do,” Gaucher said.
Short history of ASR
Doubts about ancestral sequence reconstruction -- and maximum likelihood algorithms in particular -- go far back. The idea of performing ASR first came up in 1963, but it didn’t get started until the 1990s, and back then, researchers battled fervently over wide-ranging methods.
“People would come up with the craziest notion as to why one model was best,” Gaucher said. “They’d say, ‘Well, if I simulate this weird mode of evolution along these branches here, my algorithm will work better than your algorithm.’”
The parsimony algorithm was a way of reigning in the chaos that grew out of a lack of data in evolutionary models at the time. “When the model is wrong, ‘maximum likelihood’ fails miserably,” Gaucher said.
But, now, a host of data and analysis give scientists a great picture of how evolution works (and it’s not a parsimony principle): For ages, nothing moves, then change bursts forth, then things stabilize again.
“You get this quick evolution, so lots of stuff works and lots of stuff fails, and the stuff that works then goes on and kind of maintains its status and doesn’t change,” Gaucher said. By confirming the high accuracy of the algorithms, the Georgia Tech team has also corroborated the validity of current evolutionary science they’re based on.
Kelsey Roof and Divya Natarajan of Georgia Tech coauthored the paper. Research was funded the NASA Exobiology program (grant number NNX12AI10G), DuPont (Young Professor Award) and the National Science Foundation (grant number 1145698). Any opinions, findings and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the sponsoring agencies.
Read more exciting science and technology research news at Research Horizons



Writer and contact: Ben BrumfieldResearch News(404) 660-1408